- 活動與消息
活動與消息
2018/04/09
藥事法修正新藥與學名藥品定義對生技製藥產業發展的影響
行政院2017/9/21函請立法院審議之「藥事法部分條文修正草案」,已排入4月中旬立法院審議議程。行政院在該修正草案總說明內容提到:「本次修正新藥之定義,將新興生技產品發展列為重點考量,並儘量涵蓋所有新藥型態及未來研發技術進展。又本法目前未定義學名藥及生物藥品,然藥品查驗登記審查準則已有此類產品申請查驗登記規定,經參考該準則及歐美日等先進製藥國家法令,增訂學名藥、生物藥品之定義,以符合法律明確性原則。」立法原意雖然良好,是否符合國際規範以及促進國內生技產業的發展,是本文論述的重點。
首先就此次行政院送請立法院審議的草案,有關新藥、學名藥品與生物相似性藥品引錄如下,以利本文的論述:
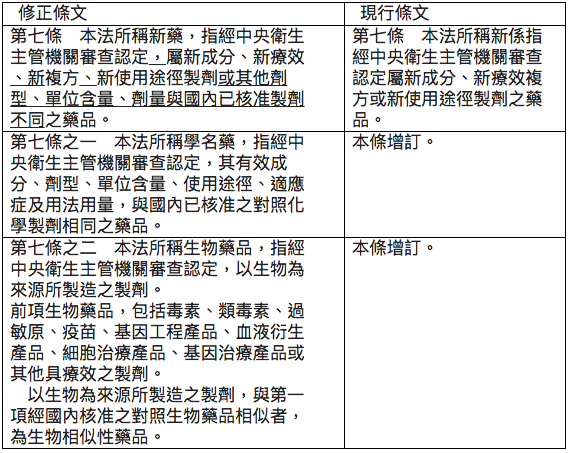
修正新藥定義對國內生技製藥產業的影響
1993年修正公布之藥事法第七條定義的新藥為「經中央衛生主管機關審查認定屬新成分、新療效複方或新使用途徑製劑之藥品」。行政院衛生署於2012/12/07修正發布藥事法施行細則第二條,將「新療效複方」定義為「指已核准藥品具有新適應症、降低副作用、改善療效強度、改善療效時間或改變使用劑量之新醫療效能,或二種以上已核准成分之複方製劑具有優於各該單一成分藥品之醫療效能者」。此種以行政裁量權將「新療效複方」拆為「新療效」與「新複方」兩大項目,不無爭議,但是在促進製藥產業發展的立意下,並未受到社會深入的檢視。
此次新藥的定義除將前述行政裁量分割之「新療效」與「新複方」入法外,再度擴大新藥定義涵蓋「或其他劑型、單位含量、劑量與國內已核准製劑不同之藥品」。這樣的定義可能為鼓勵國內藥廠發展新藥,但是此種對新藥寬鬆的定義,例如稍調整單位含量或劑量,未有創新價值的學名藥品也可符合新藥的定義,可預期以學名藥品為主的台灣製藥產業,大多數可快速升格為新藥公司,台灣將成為世界上以新藥開發為主的製藥產業國家,政府高層發展生醫產業政策目標可以提前達陣。如果政府政策的本質是僅求其「名」而非求其「實」,徒淪於與國際趨勢相違的「國王的新衣」,如此寬鬆的新藥定義應有重新審視的空間。
據筆者所聞,已有民進黨立委聯署提出對此修正條文的修正案,定義新藥為「新成分、新使用途徑、新適應症;降低副作用、改善療效強度、改善療效時間、改變使用劑量或新複方之藥品」。其雖較行政院修正草案的新藥定義較嚴謹,但亦難涵蓋國際對新藥定義的本質。如將新藥之定義為「指經中央衛生主管機關審查認定,屬新成分、新療效、新複方、新使用途徑製劑之藥品」,應可以兼顧政府政策與促進產業創新的雙重目標,立法院應廣徵各界意見審酌。
學名藥品定義入藥事法的深遠影響
國際間,各政府主管機關與製藥產業界對學名藥品的認知,主要針對原開發廠專利期滿後,其他藥廠可以向主管機關申請含有相同主成分、劑型與劑量的藥品上市。我國藥事法條文中原無學名藥品的定義,現行藥品查驗登記審查準則第四條第二款定義之學名藥品定義為: 與國內已核准之藥品具同成分、同劑型、同劑量、同療效之製劑。前三同(同成分、同劑型、同劑量)並無爭議,但是將所有學名藥品定義其「與國內已核准之藥品」(通常係指原開發廠)同療效,卻缺乏藥學科學與專業的考量。
美國FDA對於學名藥品定義為: 學名藥品為與品牌藥品(通常指原開發廠藥品) 相同劑量、安全、力價、用法、品質、表現與適應症相同的藥品(A generic drug is the same as a brand name drug in dosage, safety, strength, how it is taken, quality, performance, and intended use.) 歐盟對於學名藥品的上市許可,則要求申請廠商提供品質與對照藥品相比的人體吸收資料。顯見美國與歐盟對學名藥品上市許可的審查,均須確認其與對照藥品的品質與生體相等性。
美國食品藥物管理局(Food and Drug Administration, 簡稱FDA)出版的橘皮書(Orange Book),並未將所有核准上市的學名藥品認屬「同療效」(Therapeutic Equivalence),而是以該學名藥品申請上市審核時是否具備與對照藥品(Reference Listed Drug)藥學相等性(Pharmaceutical Equivalence, PE)與生體相等性(Bioequivalence, BE),只有兩者兼具時,FDA始給予AA學名藥品的識別碼,也就是我國現行的「同療效」;如未具備生體相等性的科學數據,只能取得AB的識別碼,第一個「A」係指藥學相等性(PE),第二個「B」則指非屬療效相等性(BE)。
我國藥品查驗登記制度自1989年才開始要求口服劑型藥品須檢附生體可用率/生體相等性(Bioavailability/Bioequivalence,也就是一般所稱的BA/BE)。簡而言之,歷年經衛福部食藥署核發的21,873張學名藥品許可證(2014年底資料),頂多三分之一是檢附BA/BE資料申請而取得許可證上市者,約三分之二的學名藥品並不具備「同療效」的認定,靜脈注射劑型的藥品因直接經由血液吸收,原則上可認定屬於「同療效」範圍。
此次藥事法修正條文草案定義,「本法所稱學名藥,指經中央衛生主管機關審查認定,其有效成分、劑型、單位含量、使用途徑、適應症及用法用量,與國內已核准之對照化學製劑相同之藥品。」檢視該定義文字,僅考慮藥學相等性(PE),對於生體相等性(BE)的規定並無著墨。
如果立法院照行政院藥事法修正條文通過學名藥品的定義,30年來中央衛生主管機關(現為食品藥物管理署)受理學名藥品查驗登記時,要求檢附生體可用率/生體相等性(BA/BE) 將乏法律依據。食藥署在缺乏法律依據下,駁回僅檢附藥學相等性(PE)的學名藥品上市申請,勢將引起諸多爭議。基於藥學科學與法律規範,建議立法院審議時,將學名藥品定義修正為: 「經中央衛生主管機關審查認定,其有效成分、劑型、單位含量、使用途徑、品質、生體吸收表現、適應症及用法用量,與國內已核准之對照化學製劑相同之藥品」。增列「品質、生體吸收表現」等文字,符合美國與歐盟的規範,也與現行我國食品署審理學名藥品查驗登記的作業相符。
審酌生物相似性藥品對照藥品的彈性
此次藥事法修正條文第七條之二第三項,增列生物相似性藥品的定義為「以生物為來源所製造之製劑,與第一項經國內核准之對照生物藥品相似者,為生物相似性藥品」。生物藥品包括毒素、類毒素、過敏原、疫苗、基因工程產品、血液衍生產品、細胞治療產品、基因治療等大分子藥品,生物藥品的製造、品質、安全與療效審查和小分子藥品差異極大,所以原廠專利期滿的生物藥品沒有「學名藥品」,而是以「生物相似性藥品」稱之。
美國FDA對生物相似性藥品的定義,規定須以FDA核准有案的生物藥品為對照藥品,這與此次藥事法增訂生物相似性藥品之定義相同;美國FDA迄今已經核准九件生物相似性藥品上市。歐盟則對生物相似性藥品之對照樣品給予若干彈性,允許經歐盟會員國之一或歐盟主管機關歐盟藥品局(European Medicine Agency, EMA)認可的生物藥品為對照藥品,不以歐盟核准者為限;歐盟對生物對照藥品(Eligibility and Reference Product)的解釋,目的在避免重複不必要的臨床試驗,允許生物相似性藥品申請廠商就某些臨床試驗/非臨床試驗數據,與尚未經歐盟核准但經EMA認可的生物對照藥品進行比較。EMA迄今已經核准39件生物相似性藥品上市,顯然歐盟對生物相似性藥品的規範教具彈性。我國衛福部食藥署目前核准上市的生物相似性藥品不到5件,落後美國、歐盟許多,也比不上鄰近的日本與韓國。除因台灣生物產業的規模與技術水準差異外,亦與我國生物相似性藥品的查驗登記規範與審核作業缺乏彈性有關。
生物相似性藥品的開發,因需進行與對照藥品的臨床試驗比對,每項生物相似性藥品開發成本為學名藥品開發成本的數十倍甚至百倍以上,從數千萬元到數億新台幣不等,比較像小分子「類新藥」的開發。對照藥品的指定或選擇,扮演生物相似性藥品開發的關鍵性樞紐。目前我國健保藥品給付項目的生物藥品僅有區區不到十五項,其健保支付費用每年已高達新台幣150億以上,超過健保每年藥品支出費用的10%以上,因此加速國產生物相似性藥品的研發與上市,不僅有利於生技製藥產業發展,增進民眾對生物藥品的可近性,對健保生物藥品費用成長有一定的節制效益。
基於以上論述,行政院藥事法修正條文第七條之二第三項生物相似性藥品之定義:「以生物為來源所製造之製劑,與第一項經國內核准之對照生物藥品相似者,為生物相似性藥品」。建議增列「或經中央衛生主管機關認定」之字眼,讓衛福部食藥署對生物相似性藥品的審核具有彈性,並符合政府發展生技製藥產業的目標,建議修正條文為:
「以生物為來源所製造之製劑,與第一項經國內核准或經中央衛生主管機關認定之對照生物藥品相似者,為生物相似性藥品。」
建構引導國家藥品政策與管理的藥事法
藥事法前身為「藥物藥商管理法」,首次立法於1970年8月,一直到1993年2月大幅修正並更名為藥事法以前,僅於1979年4月,配合藥師法的修正,將該法條文有中「藥劑師」修正為「藥師」,所有條文內容均無實質變動。
1993年之後,從行政院衛生署藥政處時期到2011衛生署藥物食品管理局成立的18年間,藥事法共修法8次;而從2011年至今,短短7年間已又歷經8次藥事法之修正,此次為TFDA成立以後藥事法第9次修正,隨著醫療器材管理專法的訂定與國家藥物查驗中心行政法人的設置,近期至少會再有兩次藥事法條文的修正。
檢視近年藥事法之修正,可歸結為藥政管理、貿易談判與促進生醫製藥產業發展等三大主軸,但是在此三大主軸之間的呼應,往往因政權更迭與主事者異動,出現斷層與前後矛盾的現象。去年,美國總統川普任命的FDA局長Scott Gottlieb,經參議院政黨壁壘分明的52:48通過認命。Gottlieb上任以來,戮力執行川普總統的藥品政策不虞餘力,川普總統的藥品政策至少包括加速創新藥品核准、鼓勵學名藥品上市與價格競爭以及鴉片類藥物濫用防制三個面向,在新藥核准時效與鼓勵學名藥品上市競爭方面,已產生立竿見影的效應。
世界各國藥物主機關的職責,在於確保上市藥物的品質、安全與療效,因屬公共衛生行政體系,其宗旨為維護與促進公共健康(Protect and Promote Public Health),我國TFDA與美國FDA皆然。美國FDA認為增進公眾對創新醫藥科技的可近性(Access to Innovation),是促進公共健康的重要元素,從而衍生出在創新醫藥科技研發過程,加強與產業互動、加速創新科技產品上市審核時效的思維。
反觀我國,生醫產業發展政策的形成,決策高層或與衛福部無直接關聯,不僅對關鍵機構衛福部食品藥物管理體制的結構與癥結缺乏瞭解,在攸關製藥產業的健保核價與藥價調控方面,衛福部健保署也是一副「帝力於我何哉」的科層慣性。檢視我國藥事法的歷次修正,受外在環境因素的驅動居多,在「維護與促進公共健康」與「發展生技製藥產業」兩大政策目標之間的連結與平衡,衛福部食品藥物管理署科層體系往往只能被動配合,難有置啄空間,只能說是「生命中難以承擔之輕」罷。
資料來源:民報 文/黃文鴻(陽明大學退休教授、曾任衛生署藥政處處長)